

ABSTRACT
Clostridium difficile infection (CDI) is a major public health threat worldwide. The use of nonsteroidal anti-inflammatory drugs (NSAIDs) is associated with enhanced susceptibility to and severity of CDI; however, the mechanisms driving this phenomenon have not been elucidated. NSAIDs alter prostaglandin (PG) metabolism by inhibiting cyclooxygenase (COX) enzymes. Here, we found that treatment with the NSAID indomethacin prior to infection altered the microbiota and dramatically increased mortality and the intestinal pathology associated with CDI in mice. We demonstrated that in C. difficile-infected animals, indomethacin treatment led to PG deregulation, an altered proinflammatory transcriptional and protein profile, and perturbed epithelial cell junctions. These effects were paralleled by increased recruitment of intestinal neutrophils and CD4+ cells and also by a perturbation of the gut microbiota. Together, these data implicate NSAIDs in the disruption of protective COX-mediated PG production during CDI, resulting in altered epithelial integrity and associated immune responses.
IMPORTANCE Clostridium difficile infection (CDI) is a spore-forming anaerobic bacterium and leading cause of antibiotic-associated colitis. Epidemiological data suggest that use of nonsteroidal anti-inflammatory drugs (NSAIDs) increases the risk for CDI in humans, a potentially important observation given the widespread use of NSAIDs. Prior studies in rodent models of CDI found that NSAID exposure following infection increases the severity of CDI, but mechanisms to explain this are lacking. Here we present new data from a mouse model of antibiotic-associated CDI suggesting that brief NSAID exposure prior to CDI increases the severity of the infectious colitis. These data shed new light on potential mechanisms linking NSAID use to worsened CDI, including drug-induced disturbances to the gut microbiome and colonic epithelial integrity. Studies were limited to a single NSAID (indomethacin), so future studies are needed to assess the generalizability of our findings and to establish a direct link to the human condition.
INTRODUCTION
Clostridium difficile is the most commonly reported nosocomial pathogen in the United States and an urgent public health threat worldwide (1). C. difficile infection (CDI) manifests as a spectrum of gastrointestinal disorders ranging from mild diarrhea to toxic megacolon and/or death, particularly in older adults (2). The primary risk factor for CDI is antibiotic treatment, which perturbs the resident gut microbiota and abolishes colonization resistance (3). However, factors other than antibiotic exposure increase the risk for CDI and the incidence of cases not associated with the use of antimicrobials has been on the rise (4). Defining mechanisms whereby nonantibiotic factors impact CDI pathogenesis promises to reveal actionable targets for preventing or treating this infection.
Recently, several previously unappreciated immune system, host, microbiota, and dietary factors have emerged as modulators of CDI severity and risk. The food additive trehalose, for example, was recently shown to increase C. difficile virulence in mice, and the widespread adoption of trehalose in food products was implicated in the emergence of hypervirulent strains of C. difficile (5). Similarly, excess dietary zinc had a profound impact on severity of C. difficile disease in mice, and high levels of zinc altered the gut microbiota and increased susceptibility to CDI (6). Importantly, there is a growing body of evidence of the essential role of the innate immune response and inflammation in both protection against and pathology of CDI (7–9). Mounting a proper and robust inflammatory response is critical for successful clearance of C. difficile, and the immune response can be a key predictor of prognosis (3, 10). In this context, specific immune mediators can facilitate both protective and pathogenic responses through the activity of molecules such as interleukin-23 (IL-23) and IL-22, and an excessive and dysregulated immune response is believed to be one of the main factors behind postinfection complications.
Epidemiological data have established an association between the use of nonsteroidal anti-inflammatory drugs (NSAIDs) and CDI (11). Muñoz-Miralles and colleagues demonstrated that the NSAID indomethacin (Indo) significantly increased the severity of CDI in antibiotic-treated mice when the NSAID was applied following inoculation and throughout the infection (12), and indomethacin exposure is associated with alterations in the structure of the intestinal microbiota (13, 14). NSAIDs are among the most highly prescribed and most widely consumed drugs in the United States (15), particularly among older adults (16), and have been implicated in causing spontaneous colitis in humans (17, 18). They act by inhibiting cyclooxygenase (COX) enzymatic activity, which prevents the generation of prostaglandins (PGs) and alters the outcome of subsequent inflammatory events. Prostaglandins, especially PGE2, are important lipid mediators that are highly abundant at sites of inflammation and infection and that support gastrointestinal homeostasis and epithelial cell (EC) health (19). NSAID use has been associated with shifts in the gut microbiota, in both rodents and humans (20–23), but these shifts have not been explored in the context of CDI.
In this report, we deployed a mouse model of antibiotic-associated CDI to examine the impact of exposure to indomethacin prior to infection with C. difficile on disease severity, immune response, intestinal epithelial integrity, and the gut microbiota. These investigations revealed that even a brief exposure to an NSAID prior to C. difficile inoculation dramatically increased CDI severity, reduced survival, and increased pathological evidence of disease. Inhibition of PG biosynthesis by indomethacin altered the cytokine response and immune cell recruitment following CDI, enhancing intestinal tissue histopathology and allowing partial systemic bacterial dissemination by dismantling intestinal epithelial tight junctions (TJs). Additionally, indomethacin treatment alone significantly perturbed the structure of the gut microbiota. These findings support epidemiological data linking NSAID use and CDI and caution against the overuse of NSAIDs in patients at high risk for C. difficile, such as older adults.
RESULTS
Indomethacin worsens C. difficile Infection in Mice and Increases Mortality.To determine the extent to which preexposure to NSAIDs influences the natural course of CDI, mice were treated with indomethacin for 2 days prior to inoculation with C. difficile (Fig. 1A). We infected C57BL/6 female mice with 1 × 104 spores of C. difficile NAP1/BI/027 strain M7404 following 5 days of pretreatment with a broad-spectrum antibiotic, cefoperazone (Fig. 1A). This brief indomethacin treatment prior to CDI dramatically decreased cecum size and increased the mortality rate from 20% to 80% (Fig. 1C) but did not significantly impact weight loss (Fig. 1D). Mice pretreated with indomethacin and infected with C. difficile also displayed histopathological evidence of more-severe cecal tissue damage compared to mice infected with C. difficile that were not exposed to the drug (Fig. 1E). Indomethacin-exposed and infected mice exhibited no change in the burden of C. difficile in the cecum (Fig. 1F), but their livers harbored significantly greater loads of mixed aerobic and anaerobic bacteria (Fig. 1G), suggesting that indomethacin pretreatment compromised intestinal barrier function during CDI and fostered microbiota translocation to the liver.

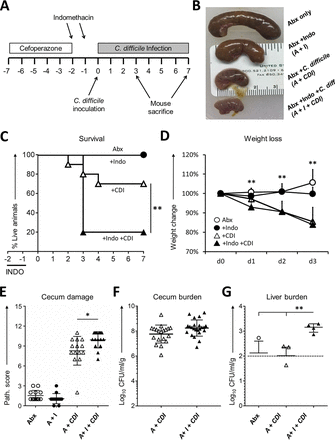
Indomethacin worsens the effects of C. difficile infection in mice. (A) C57BL/6 mice were treated with cefoperazone for 5 days followed by 2 days of recovery and then challenged by gavage with 1 × 104 spores of NAP1 strain M7404. Animals received 2 doses of 10 mg/kg of body weight of indomethacin by gavage daily as indicated by the top arrows. (B) Representative picture illustrating the macroscopic effects of the different treatments in the cecum. Indo, indomethacin; Abx, antibiotic; C. diff, C. difficile. (C to E) Mice were monitored for survival (Kaplan-Meier curve) (C), weight loss (D), and histopathologic severity of colitis (E) (n = 13 to 15/group). (F and G) C. difficile bacterial burden was evaluated in the ceca of 12 mice/group (F) and total aerobic bacterial burden plus anaerobic bacterial burden in the liver of 5 mice/group (G) also at day 3 after infection, with the discontinuous line indicating the limit of detection. Path., pathology. **, P < 0.01 (by log rank [Mantel-Cox] test for survival [panel C] and by unpaired t test for weights [panel D]); *, P < 0.05 (1-way analysis of variance [ANOVA] test for histopathological scores [panel E]); **, P < 0.01 (Wilcoxon test with Bonferroni correction [panel G]). I, indomethacin; A, antibiotic.
Indomethacin alters the proportions of neutrophils and CD4+ T cells in mucosal-associated tissues during C. difficile infection…………………………………
TO READ THE ARTICLE IN ITS ENTIRETY PLEASE CLICK ON THE FOLLOWING LINK TO BE REDIRECTED:
