Educating and Advocating for the Prevention, Treatments, Clinical Trials, Environmental Safety of Clostridioides difficile (#cdiff #CDI C. difficile) Infections Worldwide
JAMA Pediatr. Published online August 2, 2021. doi:10.1001/jamapediatrics.2021.2328
Question What is the prevalence of Clostridioides difficile detection among asymptomatic children across the age spectrum?
Findings In this systemic review and meta-analysis of 95 studies with 19 186 participants, the prevalence of detection of toxigenic and nontoxigenic C difficile was greatest (41%) among infants aged 6 to 12 months and was lowest (12%) among children aged 5 to 18 years. The prevalence of toxigenic C difficile detection was greatest (14%) among infants aged 6 to 12 months.
Meaning These findings suggest that test result interpretation should include consideration of the high likelihood of C difficile colonization in young children.
Abstract
Importance Detection of Clostridioides difficile has frequently been described in asymptomatic infants and children, but accurate estimates across the age spectrum are unavailable.
Objective To assess the prevalence of C difficile detection among asymptomatic children across the age spectrum.
Data Sources This systematic review and meta-analysis included a search of the Cochrane Central Register of Controlled Trials, MEDLINE, Embase, CINAHL, Scopus, and Web of Science for articles published from January 1, 1990, to December 31, 2020. Search terms included Clostridium difficile, Peptoclostridium difficile, Clostridioides difficile, CDF OR CDI OR c diff OR c difficile, Clostridium infections OR cd positive diarrhea OR cd positive diarrhea OR Clostridium difficile OR Peptoclostridium difficile OR pseudomembranous colitis OR pseudomembranous enterocolitis, enterocolitis, and pseudomembranous. These were combined with the following terms: bacterial colonization and colonization OR colonized OR colonizing OR epidemiology OR prevalence OR seroprevalence.
Study Selection Studies were screened independently by 2 authors. Studies were included if they reported testing for C difficile among asymptomatic children (ie, children without diarrhea) younger than 18 years.
Data Extraction and Synthesis Data were extracted independently and in duplicate by 2 reviewers. Preferred Reporting Items for a Systematic Review and Meta-analysis (PRISMA) guidelines were used. Data were pooled using a random-effects model.
Main Outcomes and Measures The primary outcome was prevalence of C difficile detection among asymptomatic children. Secondary outcomes included prevalence of toxigenic vs nontoxigenic strains of C difficile and prevalence of C difficile detection stratified by geographic region, income status, testing method, and year of testing.
Results A total of 95 studies with 19 186 participants were included. Rates of detection of toxigenic or nontoxigenic C difficile were greatest among infants aged 6 to 12 months (41%; 95% CI, 32%-50%) and decreased to 12% (95% CI, 7%-18%) among children aged 5 to 18 years. The prevalence of toxigenic C difficile colonization was lower, peaking at 14% (95% CI, 8%-21%) among infants aged 6 to 12 months and decreasing to 6% (95% CI, 2%-11%) among children older than 5 years. Although prevalence differed by geographic region (ie, North and South America vs Europe: β, −0.151, P = .001; North and South America vs Western Pacific: β, 0.136, P = .007), there was no difference by testing method (ie, culture vs polymerase chain reaction: β, 0.069, P = .052; culture vs enzyme immunoassay: β, −0.178, P = .051), income class (low-middle income vs high income: β, −0.144, P = .23; upper-middle vs high income: β, −0.020, P = .64), or period (before 1990 vs 2010-2020: β, −0.125, P = .19; 1990-1999 vs 2010-2020: β, −0.037, P = .42; 2000-2009 vs 2010-2020: β, −0.006, P = .86).
Conclusions and Relevance In this systematic review and meta-analysis, C difficile colonization rates among children were greatest at 6 to 12 months of age and decreased thereafter. These estimates may provide context for interpreting C difficile test results among young children.
To review the article in its entirety please click on the following link to be redirected. Thank you.
Sprinkle one side with salt, pepper, as desired. Spices can be omitted or replaced with spices of choice.
Place the breast in the air fryer basket seasoned side down. Then season the other side.
Cook at 360° for 9 minutes (for 8 oz chicken breast). Then flip the chicken breast over and cook for another 9 minutes. Larger chicken breasts need more cook time, and smaller ones need less time. Always use a meat thermometer to check the internal temperature.
Open the air fryer immediately so it doesn’t continue cooking in the heat.
* Food Safety: Check the internal temperature by placing the instant-read meat thermometer in the thickest part of each piece of chicken. The temperature should read 158°-160° F. If it is below that, close the lid of the air fryer to let the chicken cook in the residual heat for a few minutes (2 to 4 additional minutes). Then check it again with the meat thermometer.
Place chicken pieces on a plate and loosely cover with foil and let it rest for 5 minutes. It will continue to cook in the residual heat (and the juices will go back into the meat). Then after 5 minutes take the temperature again. The internal temperature should not be below 165° F.
Serve immediately, or prepare left-over pieces of chicken to be placed in the refrigerator immediately to be served within two days. Left-over chicken pieces can also be prepared for other recipes (e.g. chicken soup, chicken salad, chicken and pasta).
Additional Cooking Notes:
Air Fryer Chicken Breast Cook Temp & Times Turn chicken pieces over halfway through cook time (Your model and size of the air fryer may vary from this slightly)
Please check your Air Fryer Manual for cooking time and instructions.
Average Cooking Temperature and Times: 6 oz chicken breast 360° for 15 minutes 8 oz chicken breast 360° for 18 minutes 10 oz chicken breast 360° for 20 minutes
When cooking multiple chicken breasts at once, use the average weight of the chicken pieces added together.
Martinique Frentrup1, Zhemin Zhou2, Matthias Steglich1,3, Jan P. Meier-Kolthoff1, Markus Göker1, Thomas Riedel1,3, Boyke Bunk1, Cathrin Spröer1, Jörg Overmann1,3,4, Marion Blaschitz5, Alexander Indra5, Lutz von Müller6, Thomas A. Kohl7,8, Stefan Niemann7,8, Christian Seyboldt9, Frank Klawonn10,11, Nitin Kumar12, Trevor D. Lawley12, Sergio García-Fernández13,14, Rafael Cantón13,14, Rosa del Campo13,14, Ortrud Zimmermann15, Uwe Groß15, Mark Achtman2, Ulrich Nübel1,3,4
ABSTRACT
Clostridioides difficile is the primary infectious cause of antibiotic-associated diarrhea. Local transmissions and international outbreaks of this pathogen have been previously elucidated by bacterial whole-genome sequencing, but comparative genomic analyses at the global scale were hampered by the lack of specific bioinformatic tools. Here we introduce a publicly accessible database within EnteroBase (http://enterobase.warwick.ac.uk) that automatically retrieves and assembles
C. difficile short-reads from the public domain, and calls alleles for core-genome multilocus sequence typing (cgMLST). We demonstrate that comparable levels of resolution and precision are attained by EnteroBase cgMLST and single-nucleotide polymorphism analysis. EnteroBase currently contains 18 254 quality-controlled
C. difficile genomes, which have been assigned to hierarchical sets of single-linkage clusters by cgMLST distances. This hierarchical clustering is used to identify and name populations of C. difficile at all epidemiological levels, from recent transmission chains through to epidemic and endemic strains. Moreover, it puts newly collected isolates into phylogenetic and epidemiological context by identifying related strains among all previously published genome data. For example, HC2 clusters (i.e. chains of genomes with pairwise distances of up to two cgMLST alleles) were statistically associated with specific hospitals (P<10−4) or single wards (P=0.01) within hospitals, indicating they represented local transmission clusters. We also detected several HC2 clusters spanning more than one hospital that by retrospective epidemiological analysis were confirmed to be associated with inter-hospital patient transfers. In contrast, clustering at level HC150 correlated with k-mer-based classification and was largely compatible with PCR ribotyping, thus enabling comparisons to earlier surveillance data. EnteroBase enables contextual interpretation of a growing collection of assembled, quality-controlled C. difficile genome sequences and their associated metadata. Hierarchical clustering rapidly identifies database entries that are related at multiple levels of genetic distance, facilitating communication among researchers, clinicians, and public-health officials who are combatting disease caused by C. difficile.
Keywords
Clostridioides (Clostridium) difficile,
nosocomial infection,
genomic population structure,
outbreak,
cgMLST,
hierarchical clustering
Author NotesAll supporting data, code and protocols have been provided within the article or through supplementary data files. Three supplementary tables and eight supplementary figures are available with the online version of this article.Sequence accession numbers: PRJEB33768, PRJEB33779, PRJEB33780.†These authors contributed equally to this work‡These authors also contributed equally to this work
Clostridioides difficile is a major cause of healthcare-associated diarrhea and causes large infection outbreaks. Whole-genome sequencing is increasingly applied for genotyping C. difficile, with the objectives to monitor and curb the pathogen’s spread. We present a publicly accessible database for quality-controlled genome sequences from C. difficile that enables contextual interpretation of newly collected isolates by identifying related strains among published data. It also provides a nomenclature for genomic types to facilitate communication about transmission chains, epidemics, and phylogenetic lineages. Finally, we demonstrate that genome-based hierarchical clustering is largely compatible with previously used molecular typing techniques, thus enabling comparisons to earlier surveillance data.
Data Summary
All genome sequencing data were submitted to the European Nucleotide Archive (www.ebi.ac.uk/ena) under study numbers PRJEB33768, PRJEB33779 and PRJEB33780. The Clostridioides database within EnteroBase is publicly accessible at http://enterobase.warwick.ac.uk. In addition, stand-alone versions of all EnteroBase tools are available at https://github.com/zheminzhou/EToKi.
The anaerobic gut bacterium Clostridioides difficile (formerly Clostridium difficile) [1] is the primary cause of antibiotic-associated diarrhea in Europe and North America [2]. Molecular genotyping of C. difficile isolates has demonstrated international dissemination of diverse strains through healthcare systems [3–5], the community [6] and livestock production facilities [7, 8]. Previously, genotyping was commonly performed by PCR ribotyping or DNA macrorestriction. More recent publications have documented that genome-wide single-nucleotide polymorphisms (SNPs) from whole-genome sequences provide improved discrimination, and such analyses have enabled dramatic progress in our understanding of the emergence and spread of epidemic strains [9–12] and the epidemiology of local transmission [13, 14]. Eyre and colleagues have argued that transmission of C. difficile isolates within a hospital environment can be recognized with high probability as chains of genomes, which differ by up to two SNPs whereas genomes, which differ by at least ten genomic SNPs represent unrelated bacteria [13, 15]. However, SNP analyses require sophisticated bioinformatic tools and are difficult to standardize [16, 17]. A convenient alternative to SNP-based genotyping is offered by the commercial software SeqSphere, which implements a core-genome multilocus sequence typing scheme (cgMLST) for the analysis of genomic diversity in
C. difficile [18] and other organisms. Indeed, cgMLST [18] confirmed the prior conclusion from genomic SNP analyses [19] that a common clone of C. difficile had been isolated over two successive years at a hospital in China [18]. However, a recent quantitative comparison of the two methods showed that SeqSphere’s cgMLST achieved a low predictive value (41 %) for identifying isolate pairs that were closely related by the ≤2 SNPs’ criterion [20]. cgMLST of genomic sequences of a variety of bacterial pathogens can also be performed with EnteroBase (http://enterobase.warwick.ac.uk/), which has been developed over the last few years with the goal of facilitating genomic analyses by microbiologists [21]. EnteroBase automatically retrieves Illumina short-read sequences from public short-read archives. It uses a consistent assembly pipeline to automatically assemble these short-reads into draft genomes consisting of multiple contigs, and presents the assembled genomes together with their metadata for public access [22]. It also performs the same procedures on sequencing data uploaded by its registered users. Assembled genomes that pass quality control are genotyped by MLST at the levels of seven-gene MLST, ribosomal MLST (rMLST), cgMLST and whole-genome MLST (wgMLST) [21, 22]. EnteroBase supports subsequent analyses based on either SNPs or cgMLST alleles using the GrapeTree or Dendrogram visualization tools [23]. EnteroBase also assigns these genotypes to populations by hierarchical clustering (HierCC), which supports the identification of close relatives at the global level [22]. Originally, EnteroBase was restricted to the bacterial genera Salmonella, Escherichia, Yersinia and Moraxella but since January 2018, EnteroBase has included a database for genomes and their metadata for the genus Clostridioides. In June 2020, EnteroBase contained 18 254 draft genomes of C. difficile plus one genome of C. mangenotii. These included over 900 unpublished draft genomes that were sequenced at the Leibniz Institute DSMZ, as well as 80 complete genome sequences based on Pacific Biosciences plus Illumina sequencing technologies. It also included 862 unpublished draft genomes that were sequenced at the Wellcome Sanger Institute.
Here we show that comparable levels of resolution and precision are attained by EnteroBase cgMLST as by SNP analyses. We also summarize the genomic diversity that accumulated during recurring infections within single patients as well as transmission chains within individual hospitals and between neighbouring hospitals in Germany, and show that it can be detected by HierCC. We also demonstrate that HierCC can be used to identify bacterial populations at various epidemiological levels ranging from recent transmission chains through to epidemic and endemic spread, and relate these HierCC clusters to genotypes that were identified by PCR ribotyping and k-mer-based diversity analysis. These observations indicate that cgMLST and HierCC within EnteroBase can provide a common language for communications and interactions by the global community who is combatting disease caused by C. difficile.Results
Implementation of MLST schemes in EnteroBase
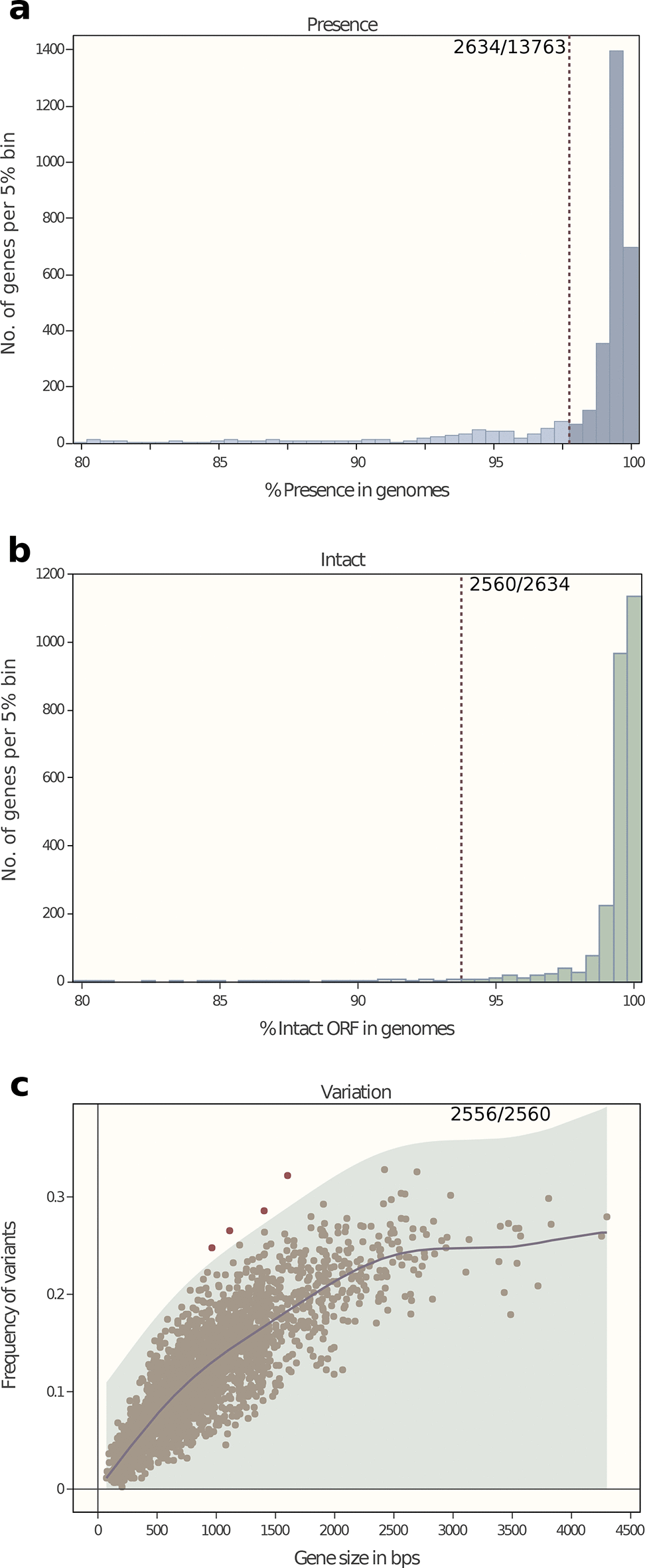
cgMLST in EnteroBase consists of a defined subset of genes within a whole-genome MLST scheme that represents all single-copy orthologues within the pan-genome of a representative set of bacterial isolates. To this end, we assembled the draft genomes of 5232 isolates of C. difficile from public short-read archives, and assigned them to ribosomal sequence types (rSTs) according to rMLST, which indexes diversity at 53 loci encoding ribosomal protein subunits on the basis of existing exemplar alleles at PubMLST [24]. We then created a reference set of 442 genomes consisting of one genome of C. mangenotii [1], 18 complete genomes from GenBank, 81 closed genomes from our work and the draft genome with the smallest number of contigs from each of the 343 rSTs (https://tinyurl.com/Cdiff-ref). The Clostridioides pan-genome was calculated with PEPPA [25] and used to define a wgMLST scheme consisting of 13 763 genetic loci (http://enterobase.warwick.ac.uk/species/clostridium/download_data). EnteroBase uses the wgMLST scheme to call loci and alleles from each assembly and extracts the allelic assignments for the subsets corresponding to cgMLST, rMLST and seven-gene MLST from those allelic calls [22]. The cgMLST subset consists of 2556 core genes, which were present in ≥98% of the reference set, intact in ≥94%, and were not excessively divergent (Fig. 1).Fig. 1.Criteria for inclusion in a cgMLST scheme of a subset of wgMLST genes based on their properties in a reference set of 442 genomes (https://tinyurl.com/Cdiff-ref). (a) Numbers of genes versus frequency (% presence) within the reference set. In total, 2634 genes satisfied the cut-off criterion of ≥98 % presence (dashed line). (b) Numbers of genes versus intact ORF (% intact ORF) within the 2634 genes from (a). Overall, 2560 genes satisfied the cut-off criterion of ≥94 % intact ORF (dashed line). (c) Frequency of allelic variants versus gene size among the 2560 genes from (b). The genetic diversity was calculated using the GaussianProcessRegressor function in the sklearn module in Python. This function calculates the Gaussian process regression of the frequency of genetic variants on gene sizes, using a linear combination of a radial basis function kernel (RBF) and a white kernel [57]. The shadowed region shows a single-tailed 99.9% confidence interval (≤3 sigma) of the prediction. Altogether, 2556 loci fell within this area and were retained for the cgMLST scheme, while four were excluded due to excessive numbers of alleles.Click to view
Comparison of cgMLST and SNPs for analyses of transmission chains
We compared the numbers of cgMLST allelic differences and the numbers of non-recombinant SNPs in isolates from multiple epidemiological chains. These included 176 isolates from four patients with recurring CDI (C. difficile infection), 63 isolates from four transmission chains in multiple hospitals [14, 19, 26], and a comprehensive sample of 1158 isolates collected over several years in four hospitals in Oxfordshire, UK [13]. A strong linear relationship (R2, 0.71–0.93) was found in all three analyses between the pairwise differences in cgMLST alleles and non-recombinant SNPs (Fig. S1, available in the online version of this article). The slope of the regression lines was close to 1.0, indicating a 1 : 1 increase in cgMLST allelic differences with numbers of SNPs. The same data were also investigated with cgMLST calculated with the commercial program SeqSphere [18], with similar correlation coefficients but a lower slope due to lesser discriminatory power of the SeqSphere cgMLST scheme (lower panels in Fig. S1).
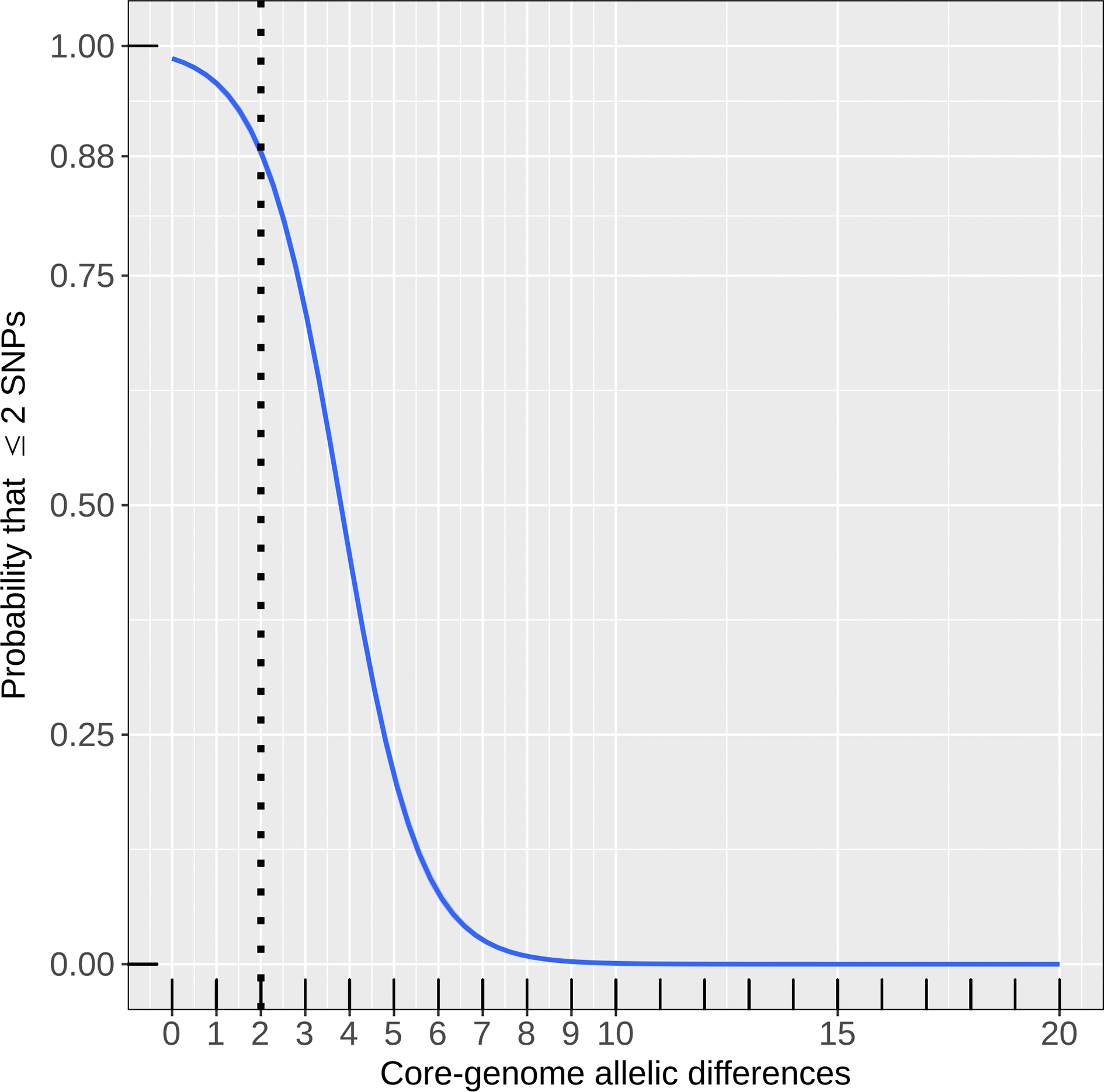
Eyre et al. [13] concluded that direct transmission between two hospital patients can be detected because their bacterial genomes differ by two SNPs or less. Our analysis indicated that these transmission chains in the Oxfordshire dataset would also have been recognized by cgMLST in EnteroBase. Genomes that differed by two cgMLST alleles usually also differed by ≤2 SNPs according to a binary logistic regression model (probability=89%; 95% confidence interval, 88–89%) (Fig. 2). Of 3807 pairs of genomes with ≤2 allelic differences, 3474 also differed by ≤2 SNPs, yielding a positive predictive value of 91 % for identifying isolate pairs with ≤2 SNPs by EnteroBase cgMLST and a sensitivity of 62 % (≤2 cgMLST allelic differences were found in 3474 of 5707 pairs with ≤2 SNPs). The comparable values for SeqSphere were 78 % positive predictive value and 99% sensitivity.Fig. 2.Binary logistic regression model to determine the probability that two genomes are related at ≤2 SNPs, given a certain difference in their cgMLST allelic profiles, based on the Oxfordshire dataset [13]. The number of SNPs was encoded as a binary dependent variable (1 if ≤2 SNPs, 0 if otherwise) and the number of allelic differences was used as a predictor variable.Click to view
We also compared the genetic distances between 242 genomes from Oxfordshire, which had been isolated during the initial 6 months and 916 genomes from the actual testing period (April 2008 to March 2011) [13]. Overall, 35% (318/916) of the latter genomes matched at least one genome collected earlier by two or less EnteroBase cgMLST alleles and 34% (316/916) matched an earlier genome by ≤2 SNPs. The two sets of genomes were 89% concordant. Thus, cgMLST is equivalent to SNP analysis for detecting inter-patient transmission chains.
Hierarchical clustering for tracing local and regional spread
SNP analyses are computer intensive, and are only feasible with limited numbers of genomes [27]. cgMLST-based relationships can be analysed for up to 100 000 genomes with GrapeTree, but analyses involving more than 10 000 genomes remain computer intensive [23]. EnteroBase implements single-linkage hierarchical clustering (HierCC V1) of cgMLST data in pairwise comparisons at multiple levels of relationship after excluding missing data [22]. These are designated as HC0 for hierarchical clusters of indistinguishable core-genome sequence types (cgSTs), HC2 for clusters with pairwise distances of up to two cgMLST alleles, etc. EnteroBase presents cluster assignments for C. difficile at the levels of HC0, HC2, HC5, HC10, HC20, HC50, HC100, HC150, HC200, HC500, HC950, HC200 and HC2500. Here we address the nature of the genetic relationships that are associated with these multiple levels of HierCC among 13 515 publicly available C. difficile genomes, and examine which levels of pairwise allelic distances correspond to epidemic outbreaks and to endemic populations.
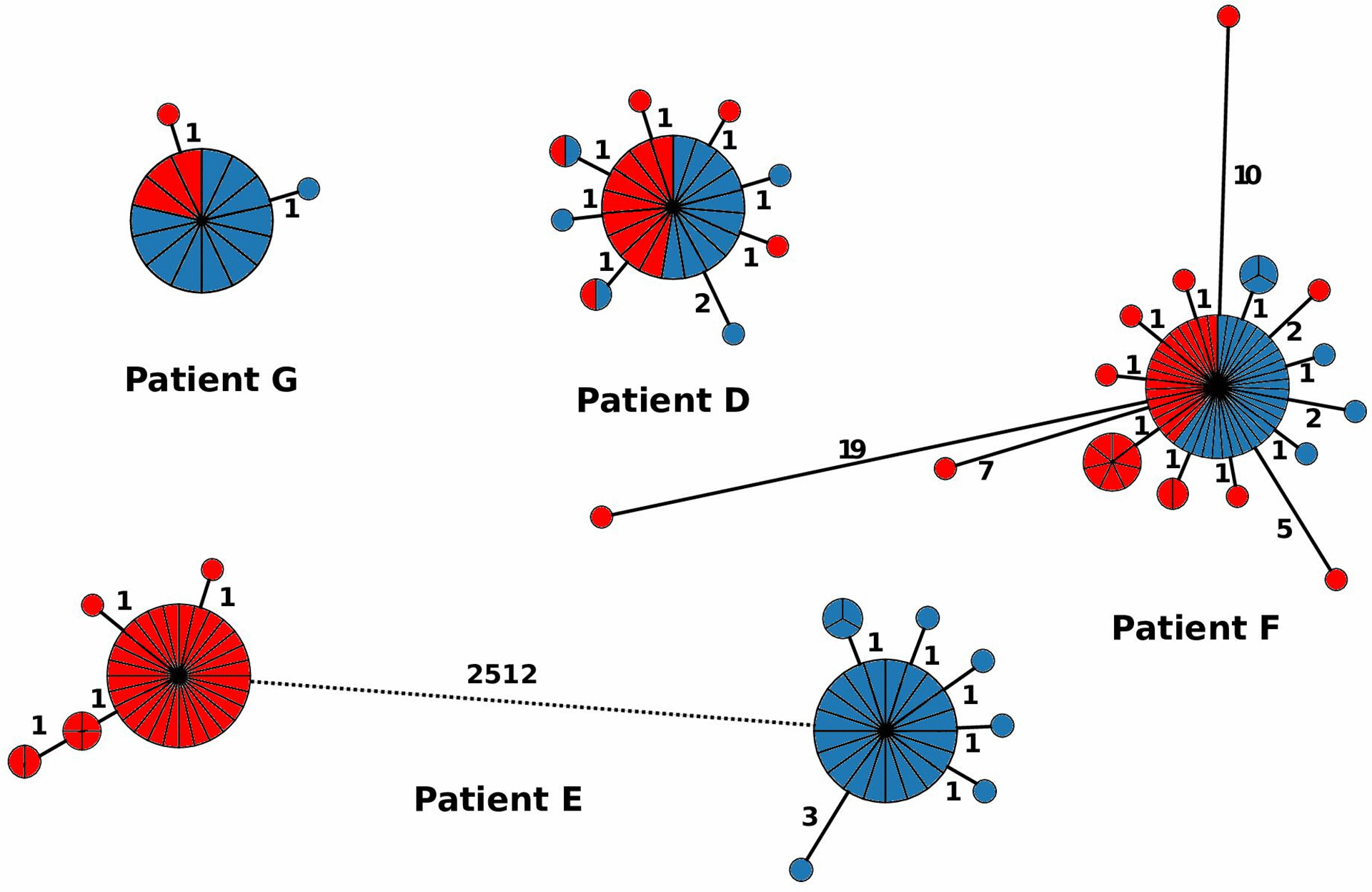
In our analyses of 176 C. difficile isolates from four patients with two recurrent episodes of CDI, multiple genomes were assigned to patient-specific HC2 clusters, some of which were isolated from the initial episode as well as the recurrence 80–153 days later (Fig. 3, patients D, F and G; 4 to 36 isolates had been collected per episode; Table S1). For these patients, relapsing disease likely reflected continued colonization after initially successful therapy. However, some isolates from patient F differed by 12–21 cgMLST alleles from the bulk population (Fig. 3), which indicates that the patient was co-infected simultaneously with multiple related strains. In patient E, the two genomes from the two CDI episodes differed by >2000 allelic differences (Fig. 3), which indicates that the second incident of CDI represented an independent infection with an unrelated strain. Hence, discrimination between relapse and reinfection based on cgMLST appears to be straightforward except that two episodes of CDI might arise by reinfection with identical strains from an environment that is heavily contaminated with C. difficile spores [28]. We note that the time intervals (16–22 weeks) investigated here exceeded the currently recommended threshold of 8 weeks for surveillance-based detection of CDI relapses [29, 30] but still yielded almost identical strains in three of four patients.Fig. 3.Minimum-spanning trees indicating the population structure of C. difficile in four patients with recurrent CDI episodes. Red, first episode; blue, second episode.Click to view
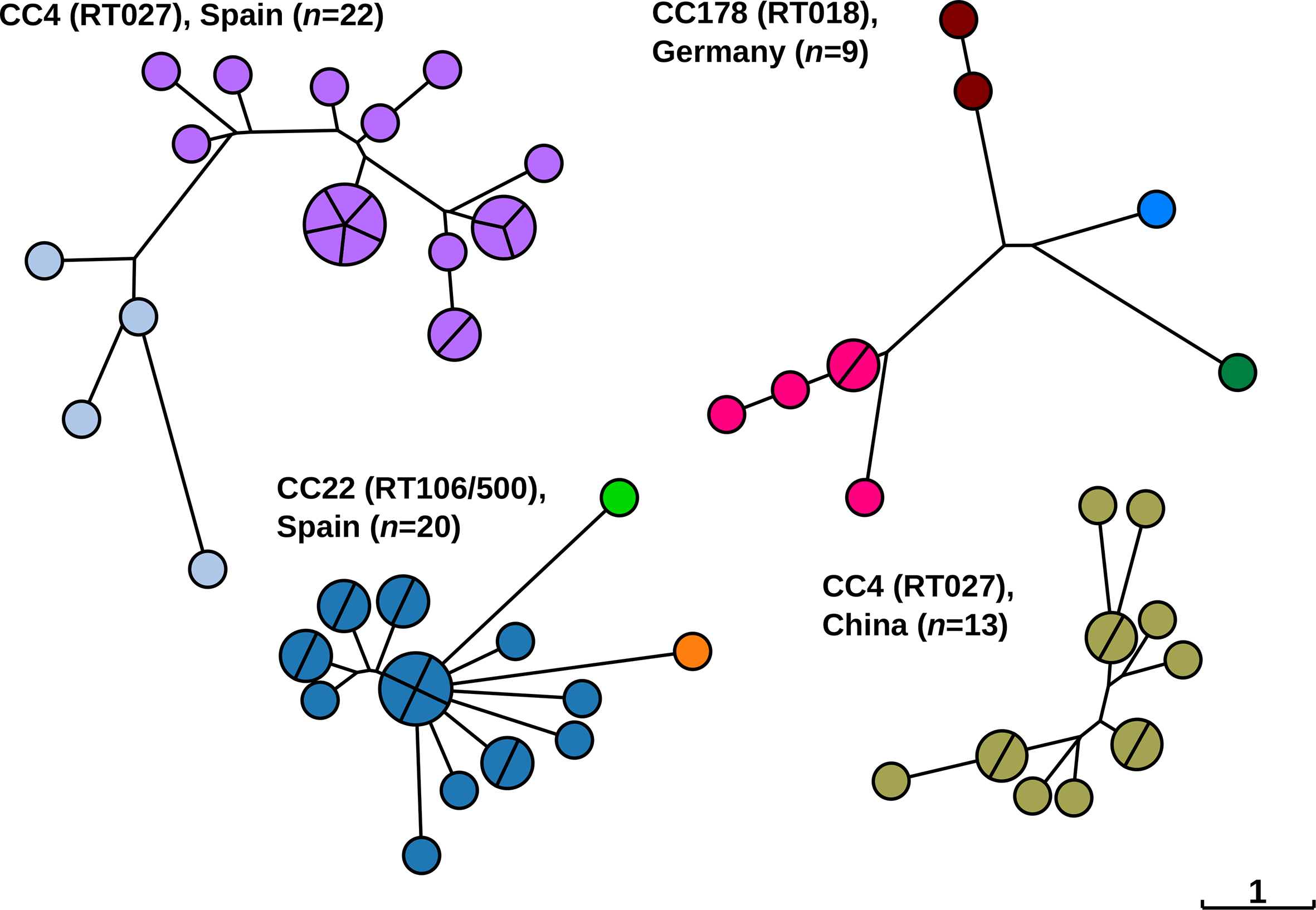
Our examinations of multiple local outbreaks have revealed individual, outbreak-specific HC2 clusters. However, it is also conceivable that multiple HC2 clusters might be isolated from a single epidemiological outbreak due to the accumulation of genetic diversity over time. Alternatively, multiple HC2 clusters within a single outbreak may represent the absence of crucial links due to incomplete sampling. Incomplete sampling of outbreaks is not unlikely because asymptomatic patients are only rarely examined for colonization with C. difficile [31–33] even though they may constitute an important reservoir for transmission. Indeed, some of the outbreaks investigated here did consist of more than one HC2 cluster (Fig. 4). For example, nine isolates from a recently reported ribotype 018 (RT018) outbreak in Germany [26] encompassed four related HC2 clusters, and outbreaks with RT027 and RT106 in a hospital in Spain [14] were each affiliated with two or three HC2 clusters (Fig. 4).Fig. 4.Neighbour-joining trees based on cgMLST showing the phylogenetic relationships among C. difficile isolates from previously published CDI outbreaks as indicated [14, 19, 26]. Nodes are coloured by HC2. CC, cgST complex, i.e. related at level HC150; RT, PCR ribotype. The scale, indicating one allelic difference, applies to all trees.Click to view
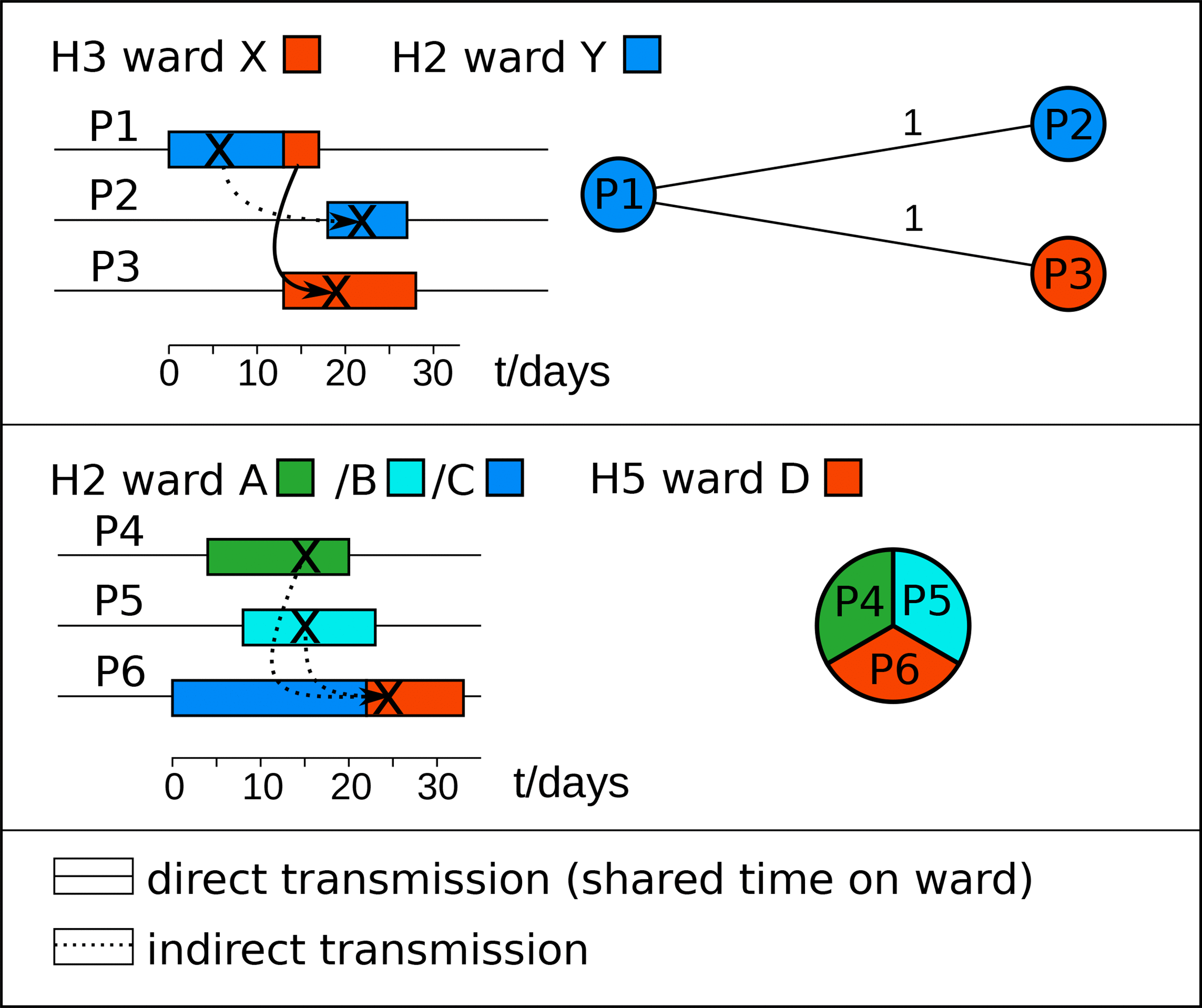
We identified 23 HC2 clusters encompassing 133 genome sequences in a dataset of 309 C. difficile genome sequences collected from CDI patients in six neighbouring hospitals in Germany. These HC2 clusters were associated with individual hospitals (Χ2, P=8.6×10−5; Shannon entropy, P=4.2×10−5) and even with single wards in these hospitals (Χ2, P=0.01; Shannon entropy, P=6.2×10−3). We investigated whether these HC2 clusters reflected the local spread of C. difficile within institutions by retrospective analyses of patient location data. Sixty-six patients (50 %) were found to have had ward contacts with another patient with the same HC2 cluster (median time interval between ward occupancy: 63 days; range, 0 to 521). These results are consistent with the direct transmission on wards of C. difficile isolates of the same HC2 cluster (Fig. 5). For patients such as P1 and P2 where the shared ward contacts were separated in time (Fig. 5), transmission may have occurred indirectly through asymptomatically colonized patients or from a common reservoir, such as environmental spore contamination [14, 31, 32]. We also detected 15 HC2 clusters that included isolates from two or more hospitals in the region. Subsequent analyses of patient location data confirmed that some of these HC2 clusters were associated with patient transferrals between the hospitals (Fig. 5). Hence, hierarchical clustering of C. difficile genome sequences in conjunction with retrospective analysis of patient movements revealed multiple likely nosocomial transmission events, none of which had been detected previously by routine surveillance.Fig. 5. Timelines of two transmission chains discovered retrospectively through inspection of files from CDI patients with closely related C. difficile isolates (HC2). Colours indicate hospital wards, ‘X’ indicates a diagnosis of CDI, and arrows indicate presumed transmission pathways. Minimum-spanning trees indicating genomic distances among C. difficile isolates are shown on the right. Upper panel: patient P1 was diagnosed with CDI in hospital H2 and transferred to hospital H3 15 days later. Another five and 6 days later, respectively, patients P2 in hospital H2 and P3 in hospital H3 got diagnosed with CDI with closely related strains. Both these patients were on the same wards as the initial patient, who probably had been the source for the pathogen. Since there was no temporal overlap between patient P2 and the other patients in hospital H2, the transmission may have occurred indirectly, possibly through environmental contamination. Lower panel: another putative transmission chain involved three patients that had shared time in hospital H2. Patients P4 and P5 got diagnosed with CDI on the same day after they had shared 7 days in this hospital, albeit on different medical wards. The third patient developed CDI with the same C. difficile cgST 4 days after being transferred to another hospital (h5), but had previously stayed at hospital H2 during the time when CDI got diagnosed in the first two patients. Since the three patients stayed on different wards in hospital H2, transmission presumably occurred indirectly.Click to view
Hierarchical clustering for identification of epidemic strains and endemic populations
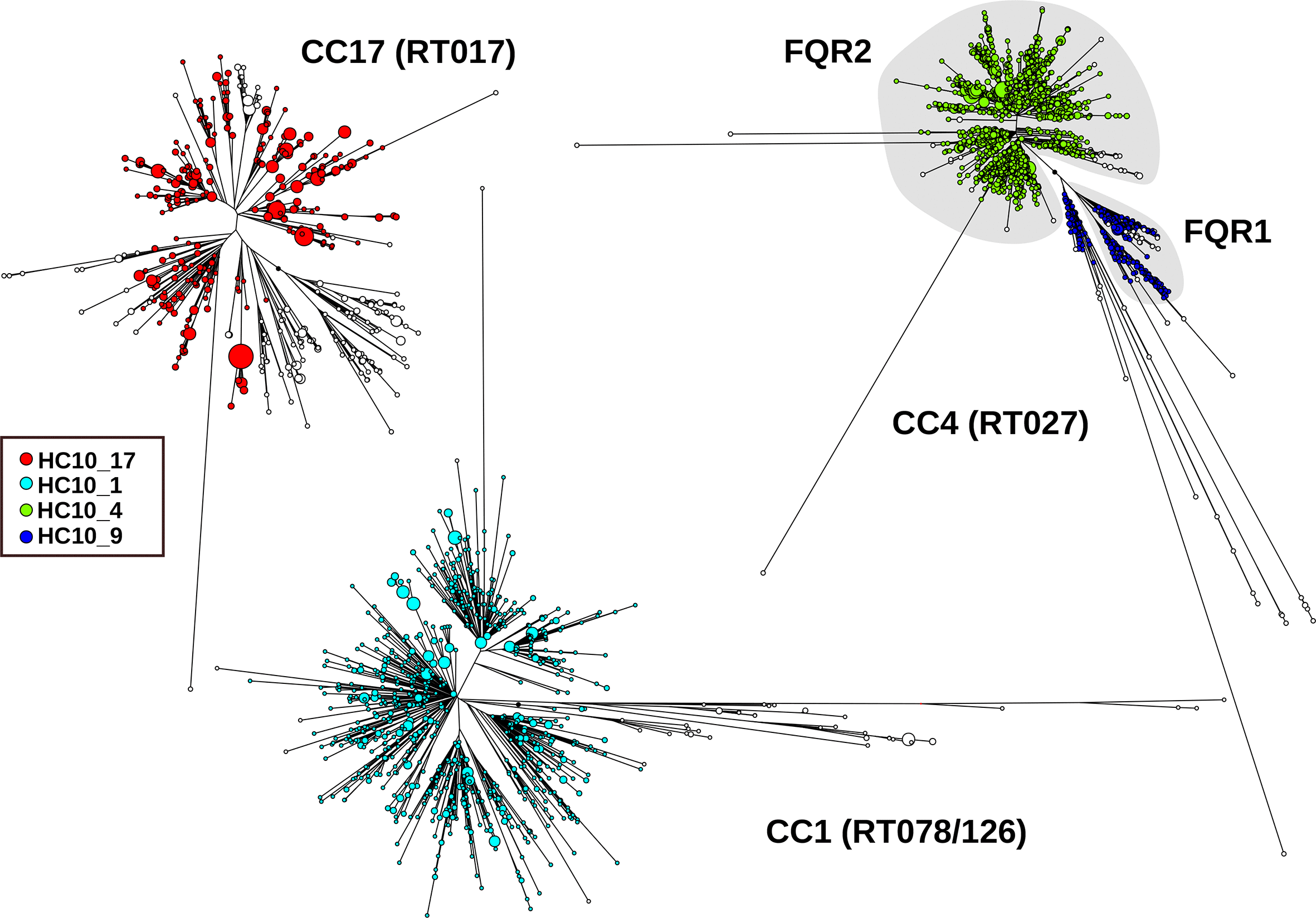
The international epidemic spread of C. difficile over up to 25 years has been inferred previously on the basis of molecular epidemiology with lower resolution techniques [34]. For multiple representatives of those epidemic strains in EnteroBase, the majority of these epidemic groups corresponded to HC10 clusters, including epidemic RT017 [11] (HC10_17), the two fluoroquinolone-resistant lineages of RT027 [9] (HC10_4, HC10_9), or livestock-associated RT078/126 [35] (HC10_1) (Fig. 6).Fig. 6.Phylogenetic structure of three international C. difficile epidemics, each of which has spread for about 25 years [9, 11]. Within each epidemic, the majority of isolates are related at level HC10, as indicated by the colours. CC, cgST complex, i.e. related at level HC150; RT, PCR ribotype.Click to view
Endemic populations have also been described by ribotyping and phylogenetic analyses, some of which have acted as sources for the emergence of epidemic strains [2, 9]. Many endemic populations seem to be represented by HC150 clusters. Clustering at HC150 was well supported statistically (Fig. S2), and the frequency distribution of pairwise genomic distances indicated that multiple database entries clustered at <150 cgMLST allelic differences (Fig. S3). HC150 clusters also correlated well with k-mer-based classification [36]. When applied to the dataset of 309 C. difficile genomes from six hospitals in Germany, the two methods implemented in EnteroBase and PopPUNK found 51 and 48 clusters, respectively, the majority of which coincided (adjusted Rand coefficient, 0.97).
A cgMLST-based phylogenetic tree of 13515 C. difficile genomes showed 201 well-separated HC150 clusters, each encompassing a set of related isolates, plus 209 singletons (Fig. 7). Because these HC150 clusters are based on cgMLST genetic distances, we refer to them as ‘cgST complexes’, abbreviated as CCs. Genomes from each of the major CCs have been collected over many years in multiple countries, indicating their long-term persistence over wide geographic ranges (Table 1).Fig. 7.Rapid-neighbour-joining phylogenetic tree based on cgMLST variation from 13 515 C. difficile genomes. Colours and numerals indicate CCs (HC150 clusters) with ≥10 entries, and information on predominant PCR ribotypes is provided in brackets.Click to view
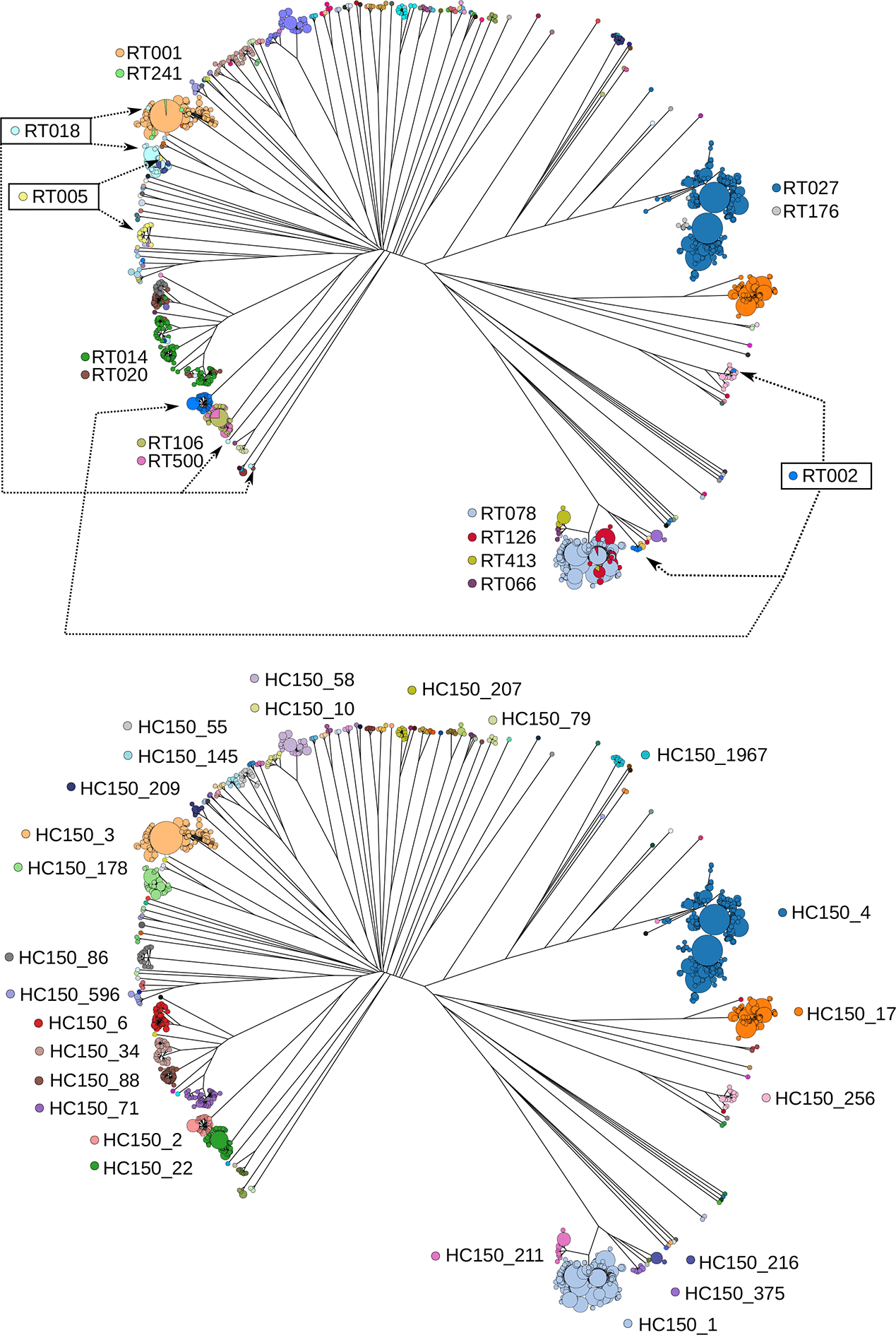
We compared HC150 clustering with PCR ribotyping for 2263 genomes spanning 84 PCR ribotypes for which PCR ribotyping data were available in EnteroBase. These included 905 genomes, which we ribotyped (Table S2), as well as several hundred other genomes for which ribotype information was manually retrieved from published data. The correlation between HC150 clustering and ribotyping was high (adjusted Rand coefficient, 0.92; 95% confidence interval, 0.90–0.93). However, our analysis also revealed that PCR ribotypes did not always correspond to phylogenetically coherent groupings. PCR ribotypes 002, 015 and 018 were each distributed across multiple phylogenetic branches (Fig. 8). Furthermore, some genomes with indistinguishable cgMLST alleles were assigned to multiple ribotypes, including RT001/RT241, RT106/RT500 and RT126/RT078 (Fig. 8, Table 1). In these cases, both ribotypes occurred in several, closely related clades (Fig. 8), indicating that similar ribotype banding patterns had evolved multiple times. In contrast, HC150 clusters corresponded to clear-cut phylogenetic groupings within a phylogenetic tree of core genes (Fig. 8b).Fig. 8.Rapid-neighbour-joining phylogenetic tree based on cgMLST variation from 2263 C. difficile genomes, for which PCR ribotyping information is available. Upper panel: nodes are coloured by PCR ribotype as indicated. Lower panel: nodes are coloured by CC (HC150 clusters).Click to view
Higher population levels
HierCC can also identify clusters at still higher taxonomic levels, up to the levels of species and sub-species [22]. In C. difficile, HC950 clusters seem to correspond to deep evolutionary branches (Fig. S4) and HC2000 clusters were congruent with the major clades reported previously [37], except that cluster HC2000_2 encompassed clade 1 plus clade 2 (Fig. S5). Finally, HC2500 may correspond to the subspecies level, because it distinguished between C. difficile and distantly related ‘cryptic clades’ (Fig. S6).Discussion
Infectious disease epidemiologists frequently seek to know if new isolates of bacterial pathogens are closely related to others from the different geographical origin, i.e. if they are part of a widespread outbreak. Unlike a previous cgMLST implementation [18], EnteroBase supports this goal by taking full advantage of rapidly growing, public repositories of short-read genome sequences [22]. In contrast to short-read archives, however, where stored sequence data are not readily interpretable without specialized bioinformatic tools [38], EnteroBase enables contextual interpretation of a growing collection (18 254 entries as of June 2020) of assembled, quality-controlled C. difficile genome sequences and their associated metadata. At least the collection date (year), the geographic origin (country), and the source (host species) are available for the majority of database entries. Importantly, phylogenetic trees based on cgMLST allelic profiles from many thousand bacterial genomes can be reconstructed within a few minutes, whereas such calculations are currently prohibitively slow based on SNP alignments [22]. Genome-sequencing reads from newly sampled C. difficile isolates can be uploaded to EnteroBase and compared to all publicly available genome data within hours, without requiring any command-line skills.
We demonstrate that the application of cgMLST to investigations of local C. difficile epidemiology yields results that are quantitatively equivalent to those from SNP analyses. This is a major advance because SNP analyses require specific bioinformatic skills and infrastructure, are time-consuming and not easily standardized [16]. A web platform for centralized, automated SNP analyses on bacterial genomes is limited to food pathogens currently and does not offer any analyses on C. difficile genomes [39]. Even though a cgMLST scheme for C. difficile had been published recently [18], its ability to identify closely related isolates and the inferred genomic distances was shown to be inferior to SNP analyses due to an excess of errors introduced by the de novo assembly of sequencing reads and a lack of per-base quality control [20]. In EnteroBase, cgMLST is also based on de novo assembly, but EnteroBase uses Pilon [40] to polish the assembled scaffolds and evaluate the reliability of consensus bases of the scaffolds, thereby achieving comparable accuracy to mapping-based SNP analyses. When applied to a large dataset of C. difficile genomes from hospital patients in the Oxfordshire region (UK), cgMLST and SNP analysis were largely consistent (89% match) at discriminating between isolates that were sufficiently closely related to have arisen during transmissions chains from others that were epidemiologically unrelated.
After assembly, draft genomes contain missing data and many cgSTs have unique cgST numbers but are identical to other cgSTs, except for missing data. Hence, individual cgST numbers are only rarely informative. However, indistinguishable cgSTs are clustered in common HierCC HC0 clusters, which ignore missing data. In June 2020, the Clostridioides database contained >12 000 HC0 clusters, indicating that the majority of genomes was unique. Similarly, EnteroBase provides cluster designations at multiple levels of HierCC, enabling rapid identification of all cgSTs that are related at multiple levels of genetic distance. The data presented here shows that HierCC designations can facilitate communications between researchers, clinicians and public-health officials about transmission chains, epidemic outbreaks, endemic populations and higher phylogenetic lineages up to the level of subspecies.
EnteroBase cgMLST identified numerous HC2 clusters of strains in C. difficile isolates that seem to have arisen during transmission chains in six neighboring hospitals in Germany. These assignments were in part consistent with retrospective investigation of patient location data, although none of the nosocomial outbreaks (defined by German law as two or more infections with likely epidemiological connections [http://www.gesetze-im-internet.de/ifsg/]) had been detected previously by standard epidemiological surveillance by skilled clinical microbiologists. Recent publications propose that prospective genome sequencing of nosocomial pathogens should be applied routinely at the hospital level to guide epidemiological surveillance [41]. Our data indicates that the combination of genome sequencing with cgMLST and HierCC may identify nosocomial transmission routes of C. difficile more effectively than presently common practice, and hence could help to reduce pathogen spread and the burden of disease. Reliable identification of transmission chains requires interpretation of pathogen genome sequence data in its epidemiological context, however [42].
HierCC will also enable comparisons to previously published data because we have provided a correspondence table between HC150 clusters and PCR ribotypes (Table 1). Rarefaction analysis indicated that the currently available genome sequences represent about two-thirds of extant HC150 (CC) diversity, which extrapolated to about 600 CCs (Fig. S7). At least some of this enormous diversity may be due to the occupation of multiple, distinct ecological niches, as exemplified by differential propensities for colonizing non-human host species (Table 1) [43, 44]. Individual CCs may also differ in their aptitudes for epidemic spread, as indicated by drastically different proportions of genomes assigned to HC2 chains: only 7% of CC141 were assigned to HC2 clusters versus 35% of CC34 and 77% of CC4 (Table 1). A full understanding of the population structure of C. difficile and its relationship to epidemiological patterns will require additional study because many of the clusters described here have not yet been studied or described. However, this task can be addressed by the global community due to the free public access to such an unprecedented amount of genomic data from this important pathogen.Methods
Sampling
In total, 309 C. difficile isolates were collected at a diagnostic laboratory providing clinical microbiology services to several hospitals in central Germany. To assemble a representative sample, we included the first 20 isolates from each of six hospitals from each of three consecutive calendar years (Table S2). For the investigation of recurrent CDI, a set of 176 C. difficile isolates were collected in a diagnostic laboratory in Saarland, Germany. Here, primary stool culture agar plates were stored at 4 °C for 5 months to eventually enable the analysis of multiple plates representing episodes of recurrent C. difficile infection from individual patients, who had developed the recurrent disease by then and could be chosen with hindsight. It was attempted to pick and cultivate as many bacterial colonies from each selected plate as possible, resulting in 6 to 36 isolates per CDI episode (Table S1). In addition, we sequenced the genomes from 383 isolates that had been characterized by PCR ribotyping previously, including 184 isolates sampled from piglets [8], 71 isolates from various hospitals in Germany [3], and 108 isolates from stool samples collected from nursing home residents (unpublished; Table S2).
PCR ribotyping
PCR ribotyping was performed as described previously [45], applying an ABI Prism 3100 apparatus for capillary electrophoresis and comparing banding patterns to the Webribo database (https://webribo.ages.at/).Whole-genome sequencing
For Illumina sequencing, genomic DNA was extracted from bacterial isolates by using the DNeasy Blood and Tissue kit (Qiagen), and libraries were prepared as described previously [46] and sequenced on an Illumina NextSeq 500 machine using a Mid-Output kit (Illumina) with 300 cycles. For generating complete genome sequences, we applied SMRT long-read sequencing on an RSII instrument (Pacific Biosciences) in combination with Illumina sequencing as reported previously [46]. All genome sequencing data were submitted to the European Nucleotide Archive (www.ebi.ac.uk/ena) under study numbers PRJEB33768, PRJEB33779 and PRJEB33780
.SNP detection and phylogenetic analysis
Sequencing reads were mapped to the reference genome sequence from C. difficile strain R20291 (sequence accession number FN545816) by using BWA-MEM and sequence variation was detected by applying VarScan2 as reported previously [46]. Sequence variation likely generated by recombination was detected through analysis with ClonalFrameML [47] and removed prior to determination of pairwise sequence distances [15] and to construction of maximum-likelihood phylogenetic trees with RAxML (version 8.2.9) [48].Genome assembly, quality control and wgMLST allele calling
Genomic data was processed by automated pipelines within EnteroBase, which were described in detail previously [22]. Briefly, Illumina sequencing reads were assembled by using Spades v3.10 [49] and assemblies were improved by applying Pilon [40]. To pass quality control, assemblies were required to comply with the following thresholds: total length, 3.6 to 4.8 Mbp; N50, ≥20 000; the number of contigs, ≤600; number of unresolved nucleotides, ≤3%; the proportion of Clostridioides sequences, >65 % (as determined by Kraken with MiniKraken database [50]). Assemblies were aligned to exemplar alleles by using blastn [51] and the usearch module UblastP [52], and allele numbers, STs, and HC numbers assigned by using the EnteroBase module MLSType [22]. All EnteroBase tools are available at https://github.com/zheminzhou/EToKi.
To determine the probability that two genomes are related at ≤2 SNPs, given a certain difference in their cgMLST allelic profiles, we inferred a logistic regression model using R ([53], pp. 593–609). Genomic relatedness was encoded as a binary response variable (1 if ≤2 SNPs, 0 if otherwise) and the number of core-genome allelic differences was used as a predictor variable. We applied this model to a dataset of 1158 genome sequences from a previous study, representing almost all symptomatic CDI patients in Oxfordshire, UK, from 2007 through 2011 [13]. While that original study had encompassed a slightly larger number of sequences, we restricted our analysis to the data (95 %) that had passed quality control as implemented in EnteroBase [21]. We used the SNP data from Eyre’s report [13].
The hierarchical single-linkage clustering of cgMLST sequence types was carried out as described [22] for all levels of allelic distances between 0 and 2556. We searched for stable levels of differentiation by HierCC according to the Silhouette index [54], a measure of uniformity of the divergence within clusters. The Silhouette index was calculated based on d^’, a normalized genetic distance between pairs of STs, which was calculated from their allelic distance d as follows: d^’=1-(1-d)^(1/l), where l is the average length (937 bp) of the genes in the cgMLST scheme.
We further evaluated the ‘stability’ of hierarchical clustering using two other criteria. The Shannon index is a measure of diversity in a given population. The Shannon index drops from nearly 1 in HC0, because most cgSTs are assigned to a unique HC0 cluster, to 0 in HC2500, which assigns all sequence types to one cluster. The gradient of the Shannon index between the two extremes reflects the frequencies of coalescence of multiple clusters at a lower HC level. Thus, the plateaus in the curve correspond to stable hierarchical levels, where the Shannon index does not change dramatically with HC level. We also evaluated the stability of hierarchical clustering by pairwise comparison of the results from different levels based on the normalized mutual information score [55] (Fig. S3).
For clustering C. difficile diversity with PopPUNK [36], we used a sketch size of 105 and a K value (maximum number of mixture components) of 15. Of note, the resulting number of clusters for the tested dataset was identical for all K between 15 and 30.
To estimate concordance between cgMLST-based hierarchical clustering and PCR ribotyping or PopPUNK clustering, respectively, we calculated the adjusted Rand coefficient [56] by using the online tool available at http://www.comparingpartitions.info/. To test statistical associations of HC2 clusters with specific hospitals and hospital wards, respectively, we compared Χ2 values and normalized Shannon entropy values (R package ‘entropy’ v.1.2.1) from contingency tables containing real isolate distributions (Table S3) and randomly permuted distributions (n=1000), by using the non-parametric, two-sided Mann–Whitney U test (R package ‘stats’ v.3.5.0).
This work was partially funded by the German Center for Infection Research (DZIF), by the Federal State of Lower Saxony (Niedersächsisches Vorab VWZN2889/3215/3266), by the EU Horizon 2020 programme (grant agreement number 643476), the Wellcome Trust (098051), and the UK Medical Research Council (PF451). EnteroBase development was funded by the BBSRC (BB/L020319/1) and the Wellcome Trust (202792/Z/16/Z), and the salary of Z.Z. was also provided by The Wellcome Trust. The funders had no role in the study design, preparation of the article, or decision to publish.
Acknowlegements:
We thank Vera Junker, Simone Severitt, Nicole Heyer and Carola Berg for excellent technical assistance, Johannes Sikorski for help with R, and David Eyre for supplying SNP data from his 2013 paper in tabular format.Author contributions
M.A. and U.N. designed the study. M.F., Z.Z., M.S., J.P.M., M.G., T.R., B.B., C.S., J.O., M.B., A.I., L.v.M., F.K., S.G.F., O.Z., U.G., M.A., and U.N. analyzed the data. L.v.M., C.S., O.Z., U.G., R.C., R.d.C., N.K., T.D.L., T.A.K. and S.N. acquired bacterial isolates and sequence data. M.F., Z.Z., M.A., and U.N. wrote the manuscript and all authors reviewed and edited the manuscript and approved the final version.
The 4th Annual Global C.diff. Awareness 2K Walks Will Now Be VIRTUAL!
Join Us On……………….
Friday, September 11th – UK
Dr. Clokie, UK Walk Event Coordinator, will be hosting the VIRTUAL Walk in Leicester on September 11th as the UK is also under strict guidelines to slow the spread of the COVID-19 virus.
The Leicester VIRTUAL Walk Will Begin at 10:00 a.m. – 11:00 a.m. – UK
VIRTUAL Entertainment Will Begin at 10:00 a.m. UK For the Children!
Saturday, September 12th – USA
The VIRTUAL Walks Will Begin at 9:00 a.m. through 12:00 p.m. EDT
VIRTUAL Entertainment Will Begin at 9:00 a.m. EDT For the Children!
All Registered Awareness Walkers Will Receive a T-Shirt, Giveaways, and More via: United States Postal Service To the Address Provided at the Time Of Registration.
To Learn More About the Global C. diff. Awareness Walk Event and How You Can Register, Please Click On the Green Button Below……………..
We are truly grateful for your efforts, support and participation of the Annual Walk events and we look forward to virtually walking with you in September!
“None of us can do this alone ~ All of us can do this together!”
~ C Diff Foundation
Clostridioides difficile in COVID-19 Patients, Detroit, Michigan, USA, March–April 2020
Avnish Sandhu, Glenn Tillotson, Jordan Polistico, Hossein Salimnia, Mara Cranis, Judy Moshos, Lori Cullen, Lavina Jabbo, Lawrence Diebel, and Teena Chopra
Author affiliations: Detroit Medical Center, Detroit, Michigan, USA (A. Sandhu, J. Polistico, H. Salimnia, M. Cranis, J. Moshos, L. Cullen, L. Jabbo, T. Chopra); Wayne State University School of Medicine, Detroit (A. Sandhu, J. Polistico, H. Salimnia, L. Diebel, T. Chopra); GST Micro LLC, Henrico, Virginia, USA (G. Tillotson)
Abstract
We describe 9 patients at a medical center in Detroit, Michigan, USA, with severe acute respiratory syndrome coronavirus 2 and Clostridioides difficile. Both infections can manifest as digestive symptoms and merit screening when assessing patients with diarrhea during the coronavirus disease pandemic. These co-infections also highlight the continued importance of antimicrobial stewardship.
Coronavirus disease (COVID-19), which is caused by infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), predominantly includes pulmonary symptoms; however, <10% of cases also include gastrointestinal events, including abdominal pain, diarrhea, and vomiting (1–4). During the COVID-19 pandemic, clinicians must be vigilant of co-infections in patients with COVID-19.
Several studies have collected data on concomitant antibiotic use in patients with COVID-19. A single-center study of 52 critically ill patients cited hospital-acquired infection in only 7 (13.5%) patients, yet 49 (94%) patients received antibiotic therapy (5). Another study, which analyzed 113 deceased patients from a cohort of 799 moderate-to-severely ill COVID-19 patients during January 13–February 12, 2020, reported that 105 (93%) deceased patients and 144 (89%) survivors had received empiric antibacterial therapy with either moxifloxacin, cefoperazone, or azithromycin (6). These antibiotics are strongly associated with C. difficile infection (CDI) (7). We report an observation of CDI as a co-occurrence or sequalae of overuse of antibiotics in COVID-19 patients.
We conducted a clinical surveillance review of CDI for all laboratory-confirmed COVID-19 patients treated at any of the hospitals belonging to Detroit Medical Center (Detroit, Michigan, USA). We screened patients by using TheraDoc software (https://www.theradoc.comExternal Link) during March 11–April 22, 2020. We abstracted data regarding baseline demographics, medical history, symptoms, laboratory values, microbiologic findings, concomitant antibiotic use, and treatment for CDI. We obtained institutional review board approval for this study.
We identified 9 cases of co-infection with SARS-CoV-2 and C. difficile. This cohort mainly included elderly patients who were predominantly female (Table). The rate of CDI at the center was 3.32/10,000 patient-days during January–February 2020 and increased to 3.6/10,000 patient-days during March–April 2020.
We noted prior CDI in 3 patients; these infections occurred 1–4 months before admission. All patients were confirmed to be positive for C. difficile by PCR and showed symptoms of diarrhea in addition to other characteristic signs and symptoms, such as abdominal pain, nausea, and vomiting. Two patients had diarrhea and were found to be positive for C. difficile at admission, whereas the remaining 7 had onset of diarrhea only after COVID-19 diagnosis; median duration from CDI diagnosis to COVID-19 diagnosis in these 7 patients was 6 days. This group of patients were severely ill, having high ATLAS scores (https://www.mdcalc.com/atlas-score-clostridium-difficile-infectionExternal Link) and multiple underlying conditions; hypertension (n = 8) and diabetes (n = 5) were the most frequent of these conditions.
Three patients received antibiotics in the month before admission; 8 received antibiotics at admission. One patient was initiated on antibiotics on day 15; this patient was also receiving antibiotics the month before admission. The most commonly administered antibiotics were cefepime (n = 5), ceftriaxone (n = 3), meropenem (n = 2), and azithromycin (n = 2). Specific CDI therapies were oral vancomycin (n = 6); vancomycin and intravenous metronidazole (n = 1); no treatment (n = 1); and a combination of oral vancomycin, intravenous metronidazole, rectal vancomycin, fidaxomicin, and fecal microbiota transplantation (n = 1). One patient who did not receive antibiotics was considered to be colonized with C. difficile. Four (44.4%) patients died during hospital admission, 1 (11.1%) was discharged to hospice, 1 (11.1%) is still hospitalized, and 3 (33.3%) were discharged to a long-term care facility.
CDI is a challenging disease, with a recurrence rate of 15%–20% and a mortality rate of 5% (8). When CDI is present as a co-infection with COVID-19, CDI therapy can be difficult to monitor if diarrhea persists because of COVID-19.
These cases highlight the importance of judicious use of antibiotics for potential secondary bacterial infection in patients with COVID-19. Antibiotics are known to have unintended consequences, such as C. difficile infection. All 9 patients received antibiotics; the median duration of antibiotic use before PCR-positive CDI was 5 days. All patients in our cohort were elderly, an age group at higher risk for complications from overuse of antibiotics, such as adverse events, antibiotic resistance, and concomitant infections like CDI (9). Secondary infections on top of CDI can increase the risk for death in patients with severe COVID-19; in this cohort, 4 patients died and 1 was discharged to hospice. To prevent CDI co-infections during the COVID-19 pandemic, integrated use of antimicrobial stewardship is needed to monitor appropriate antibiotic use.
Symptoms of CDI can complicate diagnosis of COVID-19 because both conditions can have similar manifestations; in a study of 206 COVID-19 patients, 19.4% had diarrhea as the first symptom onset (10). Of the 2 patients who had CDI diagnosed at admission, 1 patient solely had gastrointestinal symptoms, which possibly led to delayed diagnosis of COVID-19. Both COVID-19 and CDI should be considered when evaluating patients with diarrhea during the COVID-19 pandemic. Distinguishing between actual CDI versus colonization also is vital; 1 patient in our cohort was colonized. A limitation of this study is the small number of cases. However, in the face of the COVID-19 pandemic and the extensive use of antibiotics, clinicians should remain awaren of possible CDI and SARS-CoV-2 co-infection.
Dr. Sandhu is an infectious diseases–epidemiology fellow at Detroit Medical Center, Wayne State University School of Medicine. Her current research interest is in multidrug-resistant hospital-acquired infections.
Acknowledgment
G.T. is a consultant to Melinta, Crestone, Ferring, AirMmax, and Shionogi. Other authors in the manuscript have no relevant conflict of interest or financial disclosure. No funding was needed for this manuscript.
Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al.; China Medical Treatment Expert Group for Covid-19. Clinical Characteristics of Coronavirus Disease 2019 in China.N Engl J Med. 2020;382:1708–20. DOIExternal LinkPubMedExternal Link
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al.Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study.Lancet. 2020;395:507–13. DOIExternal LinkPubMedExternal Link
Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al.Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China.JAMA. 2020;323:1061–9. DOIExternal LinkPubMedExternal Link
Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, et al.Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study.Lancet Respir Med. 2020;8:475–81. DOIExternal LinkPubMedExternal Link
Chen T, Wu D, Chen H, Yan W, Yang D, Chen G, et al.Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study.BMJ. 2020;368:m1091. DOIExternal LinkPubMedExternal Link
Brown KA, Khanafer N, Daneman N, Fisman DN. Meta-analysis of antibiotics and the risk of community-associated Clostridium difficile infection.Antimicrob Agents Chemother. 2013;57:2326–32. DOIExternal LinkPubMedExternal Link
Guh AY, Mu Y, Winston LG, Johnston H, Olson D, Farley MM, et al.Emerging Infections Program Clostridioides difficile Infection Working Group. Trends in US burden of Clostridium difficile infection and outcomes.N Engl J Med. 2020;382:1320–30. DOIExternal LinkPubMedExternal Link
Biedron C, Chopra T. Issues surrounding antibiotic use in older adults.Curr Transl Geriatr Exp Gerontol Rep. 2013;2:151–8. DOIExternal Link
Han C, Duan C, Zhang S, Spiegel B, Shi H, Wang W, et al.Digestive symptoms in COVID-19 patients with mild disease severity: clinical presentation, stool viral RNA testing, and outcomes.Am J Gastroenterol. 2020;1; Epub ahead of print. DOIExternal LinkPubMedExternal Link
This website uses cookies to improve user experience. By using this website without changing the cookie settings in your web browser you consent to all cookies in accordance with the Cookie Policy.
Our Cookie Policy











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}